The Highest Quality
NeuroRestorative is committed to providing the highest quality service to those we serve. We are accredited by CARF International and the Joint Commission.


Focused on Outcomes
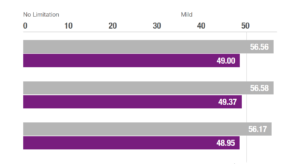
NeuroRestorative’s programs provide outcomes-oriented rehabilitation. We pride ourselves on our ability to provide the best quality services in order to achieve the best results possible.
SERVICES LOCATOR
NEWS FROM NEURORESTORATIVE
Our stories and noteworthy information from the rehabilitation field.
NewsAmerica's Most Experienced Provider
With roots dating back to 1977, NeuroRestorative is America's largest and most experienced provider of rehabilitation services for people of all ages with brain, spinal cord and medically complex injuries, illnesses and other challenges. We help people with a wide range of needs regain a productive life and a future full of promise.
Success Stories

Brady Holt and His Story of Strength
On May 14, 2016, Brady Holt, a freshman football player at Utah State University, was driving back tocollege when involved in a major motor vehicle accident that resulted in life-threatening injuries. Brady suffered a traumatic brain injury and multiple fractures, which required chest tubes, intubation and being placed on a ventilator. He was given only...
Read the Story More Stories
Our Continuum

Our continuum of care ensures ongoing focus on individuals’ well-being and progress toward the goal of a return to day-to-day living.
Neuro Institute

The NeuroRestorative Rehabilitation Learning Institute offers monthly, one hour online CEU opportunities for rehabilitation professionals. Opportunities are updated often, so make sure to check back for future presentation topics.
COVID-19 Info Center
For information and resources about vaccines, including how you can learn about eligibility in your state, please visit our COVID vaccine resource page.